
1范围
本标准规定了瓶级聚酯切片的产品分类、技术要求、试验方法、检验规则、标志、包装、运输和贮存等要求。
本标准主要适用于以精对苯二甲酸、乙二醇为主要原料, 采用直接酯化连续缩聚或间歇缩聚均聚聚酯产品和以精对苯二甲酸、乙二醇及间苯二甲酸采用直接酯化连续缩聚或间歇缩聚共聚聚酯产品。其他工艺路线的产品可参照使用。
本标准主要适用于食品容器和包装材料用聚酯切片,非食品容器及包装材料用聚酯切片可参照使用。
2引用标准
下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文。本标准出版时,所示版本均为有效,所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。
GB/T 6678—1986化工产品采样总则
GB/T 6679—1986 固体化工产品采样通则
GB/T 8170—1987 数值修约规则
GB 13114—1991食品容器及包装材料用聚对苯二甲酸乙二醇酯树脂卫生标准
GB/T 14190—1993纤维级聚酯切片分析方法 .
ISO1628—1:1998塑料 毛细管粘度计测量聚合物稀溶液的粘度第1部分:一般原理
ISO1628—5:1998塑料 毛细管粘度计测量聚合物稀溶液的粘度 第5部分:热塑性均聚和共聚聚酯
ISO3146:1985塑料半结晶聚合物熔融行为(熔融温度或熔融范围)的测定ASTM D4603:1991 聚酯特性粘度的标准试验方法
3定义
3.1色度L值:CIE标准中物质的明亮程度。
3.2色度b值:CIE标准中物质由蓝色至黄色程度。
3.3色度a值:CIE标准中物质由绿色至红色程度。
4产品分类
瓶级聚酯切片分为共聚、均聚两类,根据不同用途可分为用于矿泉水瓶、碳酸饮料瓶、其他食品容器及包装材料,按特性粘度值可分为不同规格。
5要求
5.1外观:大小均匀、呈乳白色的颗粒。无夹心、黄色及碳黑粒子和异物。
5.2卫生指标:按GB 13114规定,见附录F(标准的附录)。
5.3质量指标:见表1。
6试验方法
6.1 卫生指标
按GB 13114规定执行。
6.2质量指标
6.2.1特性粘度的测定
6.2.1.1 苯酚/邻二氯苯法(等效采用ISO 1628—1和ISO 1628—5,适用于特性粘度A的测定)
6.2.1.1.1 原理
在(25±0.05)℃的恒温水浴中分别测定溶剂和浓度为0.005 g/mL试样溶液的流出时间,计算试样的特性粘度。
6.2.1.1.2试剂
a)苯酚:分析纯;
b)邻二氯苯:分析纯。
6.2.1.1.3装置
a)恒温水浴:能控制在(25±0.05)℃范围;
b)乌氏粘度计:ISO 1628—1中的Ic型毛细管粘度计,内径φ=0.73 mm;
c)加热装置:能控制到(135-140)℃油浴或带温控加热装置;
d)磁力搅拌器;
e)不锈钢滤网:标称孔径(63~90)μm或孔径(40~100)μm烧结玻璃过滤漏斗;
f)分析天平:最小分度值为0.1 mg;
g)秒表:最小分度值为0.01 s或采用自动粘度计自动计时;
h)移液管:25 mL;
i)锥形瓶:100 mL,带磨口玻璃塞;
j)研磨机:能研磨至粒径0.5 mm左右;
k)真空干燥箱:温度控制范围:125~135℃,控制精度±5℃。
6.2.1.1.4试验步骤
a)混合溶剂:将苯酚于70℃左右的热水中溶化,苯酚和邻二氯苯按1:1的质量比充分混匀,在25℃条件下测其密度,密度控制在(1.175±0.003)g/cm3范围内,用棕色带塞玻璃试剂瓶贮存,并放置在阴暗处。最好贮藏在低温培养箱中恒温。如流出时间比配制时溶剂的流出时间差值大于1%时,则该溶剂报废。
b)测定溶剂的流出时间:将17 mL左右的苯酚一邻二氯苯混合溶剂经筛网或烧结玻璃漏斗过滤后充入乌氏粘度计中,并将其置于(25±0.05)℃的恒温水浴保持(15~20)min后,测量溶剂的流出时间,取5次测定值的算术平均值(误差不超过±0.1 s)作为溶剂的流出时间t。。
c)试样的测定:将5 g左右的样品用液氮冷却10 min,取出经粉碎机粉碎过筛后取(30~40)目的样品作为试验用样品。准确称取(0.1250±0.0001)g样品放入100 mL锥形瓶中,准确加入25 mL苯酚一邻二氯苯溶剂,在(135~140)℃的条件下不停地搅拌,使试样完全溶解,溶样时间控制在30 min内,若超过此时间,则需重新称样溶解。试样完全溶解后冷却至室温,将试样溶液经筛网或烧结玻璃漏斗过滤到乌氏粘度计中,在(25±0.05)℃的恒温水浴中放置20 min,然后测定其流出时间,重复三次,相互间的差值小于0.2 s,取三次流出时间的算术平均值作为试样溶液的流出时间。
d)粘度计的清洗:粘度计在使用前或测量值不平行时,应用浓硫酸一重铬酸钾(1:1体积比)洗液浸泡12 h以上,然后清水洗净,再用丙酮(分析纯)洗一次,在低于100℃的条件下烘干。对已使用过的粘度计清洗,可先将试液排出用三氯甲烷溶剂涮洗,然后干燥。
6.2.1.1.5结果计算
测定结果计算到小数点后四位,按GB/T 8170修约到小数点后三位。

6.2.1.2苯酚/四氯乙烷法(等效采用ASTM D 4603,适用于特性粘度B的测定)
6.2.1.2.1原理
在(25±0.05)℃的恒温水浴中分别测定溶剂和浓度为0.005 g/mL,试样溶液的流出时间,计算试样的特性粘度。
6.2.1.2.2试剂
a)苯酚:分析纯;
b)四氯乙烷:分析纯。
6.2.1.2.3装置
a)恒温水浴:能控制温度在(25±0.05)℃范围;
b)乌氏粘度计:ISO 1628—1中IB型毛细管粘度计,毛细管内径0.88 mm;
c)加热装置:能控制到(100~120)℃油浴或带温控加热装置;
d)磁力搅拌器;
e)不锈钢丝网:325目;
f)分析天平:最小分度值为0.1 mg;
g)秒表:最小分度值0.01 s或采用自动粘度计自动计时;
h)移液管:25 mL;
i)锥形瓶:100 mL,带磨口玻璃塞;
j)研磨机:能研磨至粒径0.5 mm左右;
k)真空干燥箱:温度控制在(125~130)℃,控制精度±5℃。
6.2.1.2.4试验步骤
a)配制混合溶剂:把400 g四氯乙烷加到装有600 g苯酚的试剂瓶中(称量精确至1%),在干燥箱中于60℃条件下加热,振摇至溶液完全溶解,然后将溶液通过带有槽沟过滤器放入棕色贮瓶中,放置24 h备用。溶剂密度25℃时控制在(1.232~1.238)g/cm3之间(也可按一份质量的苯酚和一份质量的四氯乙烷配制混合溶剂,25℃时溶剂密度为1.280 g/cm3)。
b)测定溶剂的流出时间:将约17 mL的苯酚-四氯乙烷溶剂经不锈钢丝网漏斗过滤后充入乌氏粘度计中并将其置于(25±0.05)℃的恒温水浴中,保持(15~20)min后,测量溶剂的流出时间,取5次测定值的算术平均值(误差不超过±0.1 s)作为溶剂的流出时间to,200 S>to>80 s。
c)试样的测定:将5 g左右的样品用液氮冷却10 min,取出经粉碎机粉碎过筛后20目的样品作为试验用样品。准确称取(0.1250土0.0001)g样品放入100 mL锥形瓶中,准确加入25 mL苯酚-四氯乙烷溶剂,在(100~120)℃的条件下加热搅拌15 min,若试样没完全溶解,继续加热到30 min,其间10 min观察一次,溶样时间控制在45 min内,若超过此时间,则需重新称样溶解。试样完全溶解后冷却至室温,将试样溶液经不锈钢丝网过滤后,充入乌氏粘度计,在(25±0.05)℃的恒温水浴中放置20 min,然后测定其流出时间,重复三次,相互间的差值小于0.2 s,取三次流出时间的算术平均值作为试样溶液的流出时间t。
6.2.1.2.5结果计算
a)由相对粘度ηr计算特性粘度[η]:

6.2.2乙醛含量的测定
6.2.2.1顶空进样法(A法)
6.2.2.1.1 原理
将粉碎后的样品在150℃条件下加热90 min,用气相色谱仪测定样品中释放出来的乙醛含量。
6.2.2.1.2试剂
a)乙醛:99%(色谱纯);
b)氮气:99.999%。
6.2.2.1.3装置
a)气相色谱仪:带FID检测器;
b)色谱工作站或积分仪;
c)顶空进样器;
d)色谱柱:不锈钢填充柱;
e)固定相:Porapak Q 80/100目;
f)带铝质盖的顶空进样器;
g)硅化聚四氟乙烯垫;
h)封盖器;
i)标准筛:孔径0.5 mm~0.6 mm(20目~30目);
j)粉碎机;
k)液氮生物容器。
6.2.2.1.4试验步骤
a)推荐的气相色谱仪操作条件:柱温:150℃;进样口温度:200℃;检测器温度:200℃。
b)求校正因子:将99%的乙醛用去氧蒸馏水配成约1 mg/mL的水溶液,并标定其浓度,见附录D。将上述乙醛水溶液用去氧水进一步稀释成浓度等距的五个标样,在顶空进样器和气相色谱仪上测定其相应的峰面积,求出校正因子。
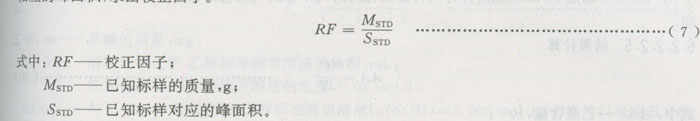
c)样品的测定:将所要测定的样品置于液氮中冷却约10 min,然后用粉碎机在液氮中将样品粉碎,粉碎时间不超过30 s,用(0.5~0.6)mm筛网过筛,取(0.5~0.6)mm的颗粒作试样。用氮气吹扫顶空瓶后,迅速称入约500 mg(精确至0.1 mg)的样品,用封盖器将事先用硅化聚四氟乙烯垫片垫好的盖子盖在顶空瓶上,将盛有试样的顶空进样器的炉体内,加热到150℃,并恒温90 min。
6.2.2.1.5结果的计算
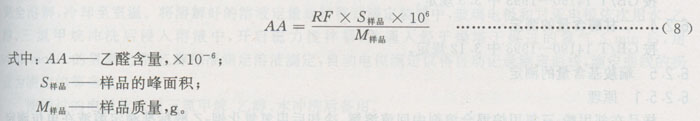
6.2.2.2裂解法(B法)
6.2.2.2.1 原理
用热分解装置将瓶级聚酯切片中含有的乙醛分离出来,用气相色谱仪通过选择适当的色谱柱和最佳测试条件达到定量测定的目的。
6.2.2.2.2试剂
a)乙醛:99%(色谱纯);
b)氮气:99.999%。
6.2.2.2.3装置
a)热分解装置;
b)气相色谱仪:与热分解装置连接的氢火焰离子化检测器;
c)记录仪或积分仪;
d)色谱柱:不锈钢填充柱(Φ3.2 mm×2 100 mm);
e)微量注射器:10μL;
f)移液管:2 mI、5 mL;
g)棕色容量瓶:1 000 mL;
h)分析天平:最小分度值为0.1 mg;
i)固定相:porapak—Q 60/80目,80/100目;GDX-102 80/100目。
6.2.2.2.4试验步骤
a)绘制标准曲线:用移液管向事先用氮气吹扫过的容量瓶中迅速加入适量的乙醛标准溶液,乙醛标准溶液的制备及分析见附录D(标准的附录),然后用蒸馏水(使用前通氮气除氧)稀释成浓度等距的五个标样,并摇匀,转入事先用氮气吹扫过的50 mL,的容量瓶中,倒满不能留有空间,放入冰箱中备用。
用微量注射器分别吸取2μL上述溶液放入裂解装置的铂舟中,推入分解管入口冷却段,转动旋塞阀,用氮气置换管中的空气5 min,再把铂舟推到加热段,在阀关闭状态下,150℃下加热10 min,然后打开阀将加热管内气体导入气相色谱仪进行气相色谱分析,记录峰面积,以乙醛含量为横坐标,峰面积为纵坐标绘制标准曲线,此标准曲线应使样品的乙醛含量落在其中部。
b)试样的测定:称取10 mg左右(准确至0.0001 g)剪碎的样品,放入裂解装置的铂舟中,推入分解管入口冷却段,转动旋塞阀,用氮气吹5 min,置换管中的空气,再把铂舟推到加热段,在阀关闭的状态下,于150℃下加热10 min,然后打开阀将加热管内的气体导入气相色谱仪中进行色谱分析,记录峰面积。
6.2.2.2.5结果计算
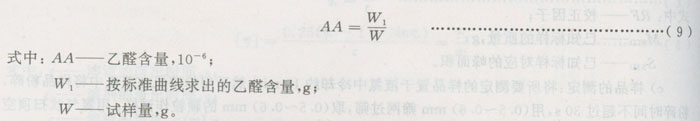
6.2.3色相的测定
按GB/T 14190—1993中3.5规定。
6.2.4二甘醇含量的测定
按GB/T 14190—1993中3.12规定。
6.2.5端羧基含量的测定
6.2.5.1 原理
样品在邻甲酚-三氯甲烷混合溶剂中回流溶解,冷却后用氢氧化钾-乙醇标准滴定溶液在电位滴定仪上测定样品中端羧基的含量。
6.2.5.2试剂
a)邻甲酚:分析纯,需经蒸馏提纯(一周内使用);
b)三氯甲烷:分析纯;
c)乙醇或苯甲醇:分析纯(苯甲醇需经蒸馏提纯);
d)草酸:分析纯;
e)氢氧化钾:分析纯。
6.2.5.3仪器
a)自动电位滴定仪:包括滴定架、玻璃电极和甘汞电极;
b)加热板:可恒温调节;
c)磨口三角烧瓶:300 mL;
d)回流冷凝管:六球以上;
e)气体洗瓶;
f)干燥塔(装有干燥剂);
g)粉碎机;
h)标准筛:20目、30目;
i)磁力搅拌器;
j)五通联接头的滴定装置(两个用于电极,两个用于氮气出、入,一个接滴定管);
k)分析天平:最小分度值0.1 mg。
6.2.5.4测试步骤
6.2.5.4.1氢氧化钾-乙醇(或苯甲醇)标准滴定溶液(约0.05 mol/L)的配制和标定
a)准确称取2.85 g氢氧化钾溶于1 000 mL乙醇或苯甲醇中。
b)精确称取(20~30)mg草酸(准确至0.1 mg),加入50 mL经煮沸后冷却的蒸馏水,溶解后进行电位滴定。
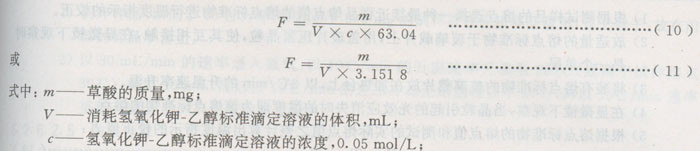
6.2.5.4.2样品测试
称取(0.5~0.6)mm粒径的样品(1±0.1)g(精确至0.000 1 g),倒入300 mL磨口三角瓶中,准确加入 50 mL邻甲酚-三氯甲烷混合溶液(7:3质量比),装上回流冷凝管,在沸腾温度下回流1 h至样品完全溶解,冷却至室温。将溶解好的溶液定量地转移到滴定装置中,玻璃电极和甘汞电极依次用水、乙醇、三氯甲烷冲洗后浸入溶液中,开启磁力搅拌器,并通入经干燥塔干燥过的氮气5 min后,用0.05 mol/L的氢氧化钾一乙醇标准滴定溶液滴定,自动电位滴定仪将自动记录滴定曲线,滴定曲线的拐点为滴定的等当点。使用过的电极依次用三氯甲烷、乙醇、水冲洗后备用。同样条件下做空白试验。
6.2.5.5结果计算

6.2.6熔点的测定
6.2.6.1 偏光显微镜法(A法,等效采用ISO 3146)
6.2.6.1.1 原理
用偏光显微镜测定瓶级聚酯切片的熔点。在控制的速率下对置于显微镜的起偏镜和检偏镜之间的试样进行加热,测定瓶级聚酯切片丧失其光学各向异性(由双折射消失得知)时的温度,以之作为熔点。
6.2.6.1.2材料
熔点标准物:试剂级,蒽:217.0℃;锡:231.9℃;二氯化锡:247.0℃;酚酞:261.8℃。
6.2.6.1.3装置
a)偏光显微镜:物镜×目镜≥50倍,带有内装检偏器的偏光凸透镜,放大倍率(×50~×100);
b)超薄切片机:薄片厚度20μm;
c)熔点测定装置:带温控装置,室温~300℃,最小分度值0.1℃;
d)玻璃载玻片:76 mm×26 mm×1 mm;
e)盖玻片:18 mm×18 mm。
6.2.6.1.4试验步骤
a)温度指示的校正
1)根据测试样品的熔点选择一种最接近样品熔点值的熔点标准物进行温度指示的校正。
2)放适量的熔点标准物于玻璃载片上,用盖玻片压紧晶粒,使其互相接触,在显微镜下观察时是一个单层。
3)将装有熔点标准物的玻璃载片放在加热台上,以2℃/min的升温速率升温。
4)在显微镜下观察,当晶粒引起的光效应消失时的温度即为该熔点标准物的熔点。
5)根据熔点标准物的熔点值和测试的实际熔点值之差计算出温度指示的校正值K:
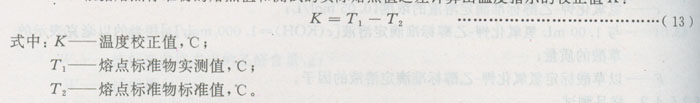
b)试样测定
1)用超薄切片机将试样切成一片厚为20μm的薄片,再用剪刀取约0.5 mm2的样品(或将试样去除表皮,切取约2~3 mg的小粒)放在玻璃载片上,用盖玻片压紧。
2)将玻璃载片放在加热台上,调整好光路,快速升温至180℃,然后以10℃/min的升温速率升温至240℃以后,再以2℃/min升温速率升温,观察初熔,记下读数(精确至0.1℃)。
3)试样达到初熔温度后,再快速升温至280℃,使其在此温度下保持3 min,然后冷至室温,再以10℃/min的升温速率升温至240℃后,以2℃/min升温速率升温,观察晶粒引起的光效应消失时的温度,记为丁(精确至0.1℃)。
6.2.6.1.5结果计算
6.2.6.1.6允差
同一样品测定两次的平行样误差应小于1.0℃。
6.2.6.2热分析法(B法,等效采用ISO 3146)
6.2.6.2.1 原理
在一个适用的DTA或DSC仪器上,按控制的速率加热一个瓶级聚酯切片试样和一个适宜的参照材料。由记录下来的DTA或DSC曲线确定与熔化性能有关的一个或几个特征点。
6.2.6.2.2材料
熔点标准物:铟、锡。
6.2.6.2.3装置
差热分析仪或示差扫描量热仪。
6.2.6.2.4试验步骤
a)温度指示的校正
1)称取5.0 mg的铟放在铝质样品盘里盖上盖子,放在前面的样品平台上,用空的盖上盖子的样品盘作为参比,放在后面的样品平台上。以30 mL/min的速率通入氮气,10℃/min的升温速率将温度从130℃程序升温至175℃,并记录下这个过程,重复上述步骤,计算二次实测温度的平均值,设为T1。输入T1值和铟的熔点标准值,重复上述步骤进行校正。
2)用锡来代替铟,重复上述步骤,以10℃/min的升温速率将温度从210℃程序升温至255℃,并记录下这个过程,重复上述步骤,计算二次实测温度的平均值,设为T20'输入T2值和锡的熔点标准值,重复上述步骤进行校正。
b)试样熔点的测定
1)粉碎样品,以低于60目为好,称取小于5.0 mg的试样放在铝质样品盘里,盖上盖子,放在前面的样品平台上。
2)以30 mL/min的速率通入氮气,以10℃/min的升温速率升温至280℃(或高于试样熔点30℃),保温10 min,然后以10℃/min的速率冷却至结晶峰以下50℃,再以10℃/min速率重新升温至280℃,记录加热曲线。
6.2.6.2.5结果计算
从曲线上读取试样的熔点值,精确至小数点后一位,按GB/T 8170修约到整数位。
6.2.7粉屑的测定
按GB/T 14190-1993中3.8规定。
6.2.8水分的测定
按GB/T 14190—1993中3.7规定。
6.2.9结晶度的测定
6.2.9.1 密度法
6.2.9.1.1原理
在装有密度梯度溶液的玻璃管中,根据切片粒子在密度梯度溶液的不同位置,测定出切片的结晶度。
6.2.9.1.2试剂
a)正庚烷:分析纯;
b)四氯化碳:分析纯。
6.2.9.1.3装置
a)结晶度测定仪;
b)密度梯度管:高度700 mm左右,直径(70~80)mm;
c)标准密度小球:测量范围(1.340~1.400)g/cm3的不同的五只小球;
d)毛细管漏斗:内径0.5 mm左右,长度大于700 mm;
e)磁力搅拌器。
6.2.9.1.4试验步骤
a)密度梯度溶液的配制:A法和B法任选一种。
1)A法(间歇法)
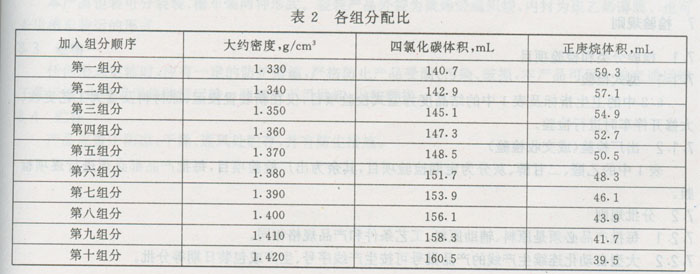
按表2要求配制200 mL不同组分的溶液,经充分搅匀后,用毛细管漏斗按组分顺序慢慢地往密度梯度管内加入,待全部加完后,用毛细管漏斗在溶液中横向轻轻搅拌数次,注意不能破坏溶液密度梯度的分布。
2)B法(连续法)
在一只2 L的三角瓶中分别加入正庚烷和四氯化碳,配制成密度为1.330 g/cm3的溶液,将此溶液加入到一只连有毛细管的三角瓶中;在另一只两升的三角瓶中加入一定量的四氯化碳(密度为1.595 g/cm3),把它和上述连有毛细管的三角瓶用乳胶管连接(见图1),并使两瓶溶液液面处于同一水平面;先打开高密度溶液瓶处的控制开关,再打开低密度溶液瓶处的控制开关,使混合溶液沿毛细管漏斗缓慢流入到密度梯度管中。

准确配制一管密度梯度液的时间为2~4 h。
b)往密度梯度管中放入五只标准密度小球,以小球的密度值作为纵坐标,小球所处的高度值为横坐标,绘制密度(g/cm3)-高度(mm)标准曲线,每次测试前,重新校准和制作标准曲线。
c)样品分析:将切片投入到密度梯度管的溶液中,放切片时要尽量轻缓,以免破坏溶液的梯度。待切片位置不再变化时读取样品聚集最多处的高度(mm),根据高度在密度一高度标准曲线上查得试样的密度。每个样品测定一次,每次测试3~30粒切片。
6.2.9.1.5结果的计算

6.2.9.2密度天平法
见附录E。
6.2.10灰分的测定
按GB/T 14190-1993中3.10规定。
7检验规则
7.1检验分类和检验项目
7.1.1型式检验
5.2中的卫生指标及表1中的结晶度为型式检验项目,仅在新装置投运、原材料及重大工艺更改、大修开停车时进行检验。
7.1.2出厂检验(或交收检验)
表1中的乙醛、二甘醇、灰分为定期检验项目,其余为出厂检验项目,每批产品都必须进行逐项检验。
7.2分批规则
7.2.1 每批产品必须是原料、辅助原料、工艺条件和产品规格相同。
7.2.2大型自动化连续生产线的产品批号可按生产线序号、生产或包装日期等分批。